

Famílias carentes, geralmente desassistidas pelo poder público e relegadas a uma parte invisível da saúde no Brasil: assim o pesquisador do Instituto de Biociências da USP, Allysson Allan de Farias, descreve as populações nordestinas que apresentam elevadas taxas de consanguinidade. Esse tipo de união, em que os membros do casal têm algum grau de parentesco, contribui para o aumento do número de indivíduos com deficiências provocadas por doenças genéticas na região.
Em sua tese de doutorado, A ancestralidade de populações do Nordeste brasileiro com elevadas frequências de casamentos consanguíneos e prevalência de doenças genéticas raras, defendida em setembro de 2018, Farias buscou identificar quando e onde surgiram as mutações que causam as chamadas doenças de herança recessiva no interior nordestino. Parte inicial do estudo foi aprovada para publicação na revista Scientific Reports com o título “Origin and age of the causative mutations in KLC2, IMPA1, MED25 and WNT7A unravelled through Brazilian admixed populations” (Origem e idade das mutações causais em KLC2, IMPA1, MED25 e WNT7A foram reveladas em populações brasileiras miscigenadas).
A tese faz parte de um projeto desenvolvido desde 2005 pela parceria entre o Centro de Pesquisa sobre o Genoma Humano e Células-Tronco (Instituto de Biociências-USP) e a Universidade Estadual da Paraíba (UEPB), cujo objetivo é identificar e estudar as novas doenças genéticas decorrentes de uniões consanguíneas nas famílias do nordeste. Como esse tipo de vínculo afetivo ocorre por várias gerações em algumas comunidades da região, observa-se um efeito cumulativo de compartilhamento de alelos – genes responsáveis pela determinação das características biológicas dos seres – iguais entre os indivíduos, condição conhecida como homozigose, que, por sua vez, favorece o aumento da frequência de pessoas afetadas por doenças genéticas de herança recessiva.
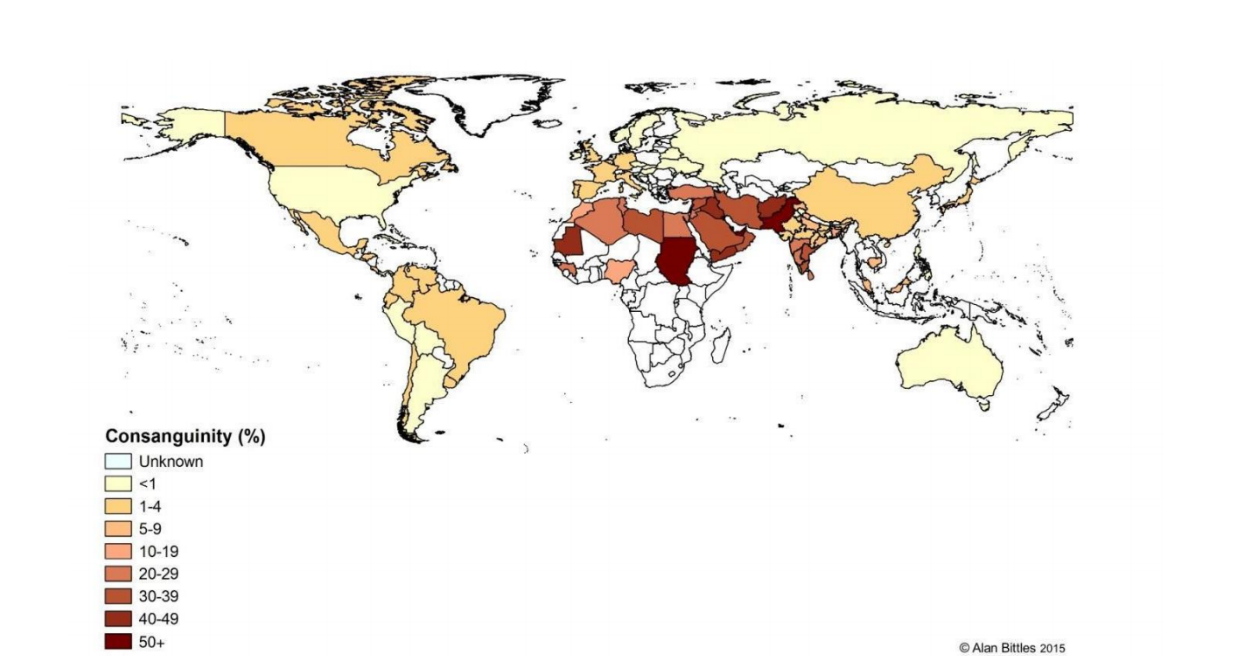
Uma das patologias descritas pelo grupo foi a síndrome Spoan, que afeta mais de 70 indivíduos – todos filhos de casais aparentados – com ancestrais originários do interior do Rio Grande do Norte e da Paraíba. Além desses estados, também foram identificados mais dois irmãos com a síndrome no Rio Grande do Sul, uma paciente originária de Minas Gerais e, em 2015, dois irmãos egípcios, todos frutos de uniões consanguíneas. “A partir da descoberta desse caso no Egito, ficamos pensando: como é que a mesma síndrome, com a mesma mutação, foi aparecer tanto nesses irmãos egípcios quanto nessa população do nordeste brasileiro?”, conta Farias.
A síndrome Spoan é ocasionada pela remoção de um segmento de cromossomo – estrutura formada por uma molécula de DNA associada a moléculas proteicas– na região não codificadora de proteínas do gene KLC2, encontrada em alta frequência nas famílias nordestinas que compartilham os mesmos alelos recessivos. Seus sintomas associam presença de rigidez muscular e dificuldade de movimento (paraplegia espástica), disfunção de um ou mais nervos periféricos (neuropatia) e perda de fibras do nervo óptico que reduzem o campo de visão (atrofia óptica).
Além da Spoan, mais duas doenças genéticas de herança recessiva que causam deficiência intelectual foram identificadas em outras duas famílias, uma de Brejo dos Santos e outra de Catolé do Rocha, na Paraíba. A primeira é causada por mutação simultânea em dois alelos iguais (mutação bialélica) no gene IMPA1, enquanto a segunda associa-se a mutação bialélica no gene MED25. No município de Riacho de Santana, Rio Grande do Norte, ademais, foi encontrada uma família com seis indivíduos que apresentavam atrofia de um órgão ou tecido por parada do desenvolvimento na fase embrionária – agenesia ou hipoplasia de fíbula – com outras anomalias de membros provocadas por mutação no gene WNT7A, condição que, em 2008, recebeu o nome de síndrome Santos.
A partir desse cenário, o pesquisador investigou a origem e a idade das mutações dos genes responsáveis por essas quatro doenças genéticas observadas na região. “Fizemos um estudo de ancestralidade para ver as contribuições africanas, nativo-americanas e europeias para os genes KLC2, IMPA1, MED25 e WNT7A contendo mutações, considerando que a população brasileira é tri-híbrida”, explica.
A pesquisa identificou que três dessas doenças genéticas raras têm um haplótipo – definido por Farias como “blocos de cromossomos que são quebrados por recombinação e passam de uma geração para outra” – europeu: a síndrome Spoan e as dos genes IMPA1 e MED25. A síndrome Santos, ocasionada por mutação no gene WNT7A, por sua vez, apresentou haplótipo nativo-americano.
Farias conta que esses resultados originaram duas novas questões ao estudo: quando foi o surgimento das doenças de haplótipo europeu e se a origem daquela com haplótipo nativo-americano ocorreu antes ou depois do contato com os portugueses.
“Vimos que a síndrome Spoan, que apresenta essa conexão Egito-Brasil, tem mais ou menos 485 anos, ela recupera a história do início da entrada de portugueses na costa do Brasil e de grandes migrações que estavam ocorrendo na região da Península Ibérica e norte da África”, explica.
As doenças ligadas aos genes IMPA1 e MED25 apresentaram idade de 200 anos, e a Síndrome Santos, provocada por mutação no gene WNT7A, por volta de 100 anos; as três foram classificadas pelo pesquisador como relativamente recentes. “Felizmente, a síndrome Santos tem uma idade muito recente, surgiu depois do contato com os portugueses e não teve origem dentro das populações indígenas que habitaram aqui de 10 mil anos para cá, como se pensava”, esclarece.
Na segunda parte do estudo, ainda, o pesquisador buscou determinar a origem da linhagem materna das famílias dos indivíduos com mutação em KLC2, ou seja, dos portadores da síndrome Spoan. A matrilinhagem exibiu evidências de ligação entre as mães de famílias de pessoas com mutação no gene e mães de judeus sefarditas – termo que remete aos descendentes de judeus originários de Portugal e Espanha.
Foi encontrado, então, um possível grupo de haplótipos fundador que pode reforçar narrativas históricas de entrada de mães sefarditas no litoral do Nordeste junto com os holandeses. “Associada ao estudo da idade da mutação, a análise da matrilinhagem fez com que recuperássemos a história de mulheres e mães que adentraram no interior do nordeste brasileiro provavelmente porque se refugiavam da Inquisição de Portugal e da Espanha, entre 1630 e 1654, que foi o período em que os holandeses dominaram toda a costa do nordeste do país e facilitaram a entrada e saída de judeus”, conta.
A pesquisa de Farias sugere que as práticas atuais de casamentos consanguíneos no nordeste do país poderiam estar relacionadas a traços culturais deixados pela atuação da Inquisição de 300 a 400 anos atrás. Isso, pois os “cristãos velhos” (grupos de católicos-portugueses) e os “cristãos-novos” (judeus convertidos ao catolicismo) respeitavam os estatutos de sangue baseando-se em ordens da Inquisição que proibiam casamentos entre católicos e judeus portugueses.
“Minha pesquisa procura fazer esse resgate histórico de doenças genéticas que acometem essas comunidades nordestinas, valorizando os indivíduos e mostrando um pouco do sofrimento dessas famílias e das genealogias complexas do interior do Rio Grande do Norte e da Paraíba”, considera.



Faça um comentário